Bedtools tutorial
Help 13 / 20
bedtools genomecov
Measure genome-wide coverage
For many analyses, one wants to measure the genome wide coverage of a feature file. For example, we often want to know what fraction of the genome is covered by 1 feature, 2 features, 3 features, etc. This is frequently crucial when assessing the “uniformity” of coverage from whole-genome sequencing. This is done with the versatile genomecov tool.
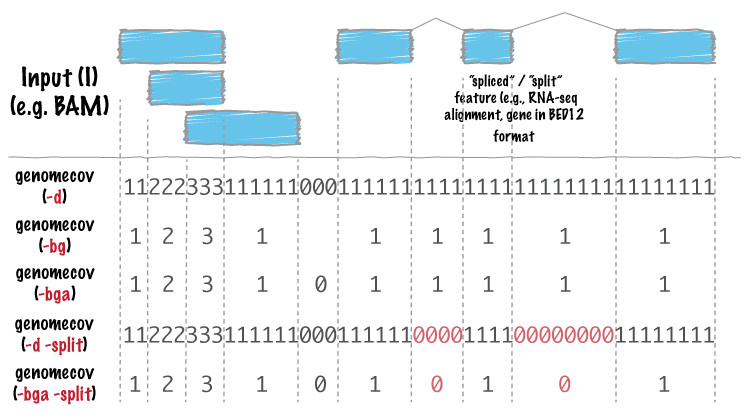
As an example, let’s produce a histogram of coverage of the exons throughout the genome. Like the merge tool, genomecov requires pre-sorted data. It also needs a genome file as above.
bedtools genomecov -i exons.bed -g genome.txt
Using the -bg option, one can also produce BEDGRAPH output which represents the “depth” fo feature coverage for each base pair in the genome:
bedtools genomecov -i exons.bed -g genome.txt -bg | head -n 20
Loading...