Bedtools tutorial
Help 9 / 20
bedtools merge
Merge regions
Many datasets of genomic features have many individual features that overlap one another (e.g. alignments from a ChiP seq experiment). It is often useful to just combine the overlapping into a single, contiguous interval. The bedtools merge command will do this for you.
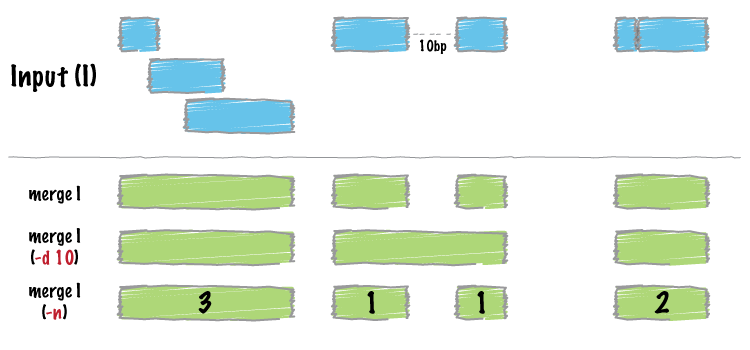
The merge tool requires that the input file is sorted by chromosome, then by start position. This allows the merging algorithm to work very quickly without requiring any RAM. If your files are unsorted, the merge tool will raise an error. To correct this, you need to sort your BED using the UNIX sort utility. For example: sort -k1,1 -k2,2n foo.bed > foo.sort.bed
Loading...